e:Bio Cellemental rxncon : The signal transduction network of baker’s yeast
We are working towards comprehensive mechanistic models of the regulatory networks that cells use to take decisions.
The cellular regulatory networks monitor the state of a cell and its surroundings, and control key cellular processes such as metabolism, cell division and apoptosis. One of the main challenges of systems biology is to provide a mechanistic (as opposed to phenomenological) understanding of these networks in terms of their elementary building blocks: the reactions between and states of biological molecules.
We are inspired by the comprehensive wall charts and genome-scale models that revolutionised our understanding of cellular metabolism. However, these methods do not work well for signal transduction, primarily due to issues with scalability.
Scalability is a fundamental problem in the description of cellular networks, in particular when aiming for genome-scale models (GSMs). The actual problem is twofold, first in the model formulation, and second in the model execution: even when the formulation of a model does not run into scalability issues, the execution or simulation might still be infeasible.
This issue is most pronounced for the cellular regulatory networks that process information. Information is encoded in site specific states of components (e.g. modification at specific residue, ligand binding at specific domain), and that most of these state changes can be combined. While not an issue in small networks, the combinatorial complexity increase rapidly forcing heavy handed simplification in even small pathway models and precluding the development of GSMs for regulatory networks.
To solve these issues, we develop rxncon (“reaction-con”), the reaction-contingency language and toolbox, and we just released rxncon 2.0, the second generation rxncon language.
The rxncon language describes signal transduction at the same abstraction level as empirical data, and hence minimise both the (unnecessary) complexity and assumptions. With rxncon, we can build, visualize and simulate large mechanistic models of signal transduction using a higher level biological language that can be compiled into graphical or executable formats.
We are now using rxncon to build comprehensive - in scope and detail - models of signal transduction in yeast, human and plant cells. For further information and literature, please refer to https://rxncon.org.
Scalable modeling
The rxncon language
The first and arguably most important step of modeling is to formalize biological knowledge. With rxncon, we develop a natural language that captures the molecular biology of cellular signal transduction at the same resolution that it is measured: In terms of elemental states.
Elemental states are site-specific states, e.g. (the lack of) a specific modification at a specific residue or a bond between two specific components. A components exact configuration, or microstate, is often defined by several elemental states - which are rarely measured simultaneously (at least not in the same molecule). Hence, inferring microstates from elemental states is problematic for two reasons: First, the number of microstates increases exponentially with the number of elemental states, leading to the well-known combinatorial complexity of signaling. Second, the microstates have not been measured, introducing an ambiguity between data and model. Both can be avoided by expressing the model in terms of elemental states.
The key invention in the rxncon language is to use a bipartite network definition, consisting of elemental reactions and contingencies.
The elemental reactions are minimal and decontextualized reaction events, which states a possibility (e.g. that a specific kinase can phosphorylate a target on a specific residue) but no contextual information. The contingencies are statements on which (combination of) elemental states are necessary for an elemental reaction to fire (e.g. that, for a kinase to phosphorylate a target on a specific residue, the kinase must be phosphorylated on a specific residue in its activation loop (and hence active)). Both reactions and contingencies are expressed in terms of elemental states, and together they define a specific reaction network.
A carefully built rxncon network constitutes a detailed knowledge base that can be compiled into graphical or executable output formats.
Visualization
Visualization is a powerful tool in model construction, debugging and analysis. We develop three visualization methods for rxncon network: The reaction graph, the elemental species-reaction graph and the regulatory graph.
For a model to mechanistically explain signal transduction, the network must be connected at two levels: First, there must be a chain of reactions connecting the most upstream and downstream components (e.g. a phosphorylation cascade). Secondly, the outcome of each reaction must trigger a change in downstream reaction rates.
The reaction graph visualizes the reaction layer. The graph contains components, domains and residues (and nodes) that connected by elemental reactions (edges). This high-level graph can be used to visualize reaction chains or high throughput or genetic data (which typically are measured on the level of components).
The elemental species-reaction graph visualizes the complete rxncon model. This bipartite graph contains elemental reactions and states (as nodes) and reaction changes and contingencies (as edges). It visualizes how elemental reactions synthesize, degrade, produce or consume elemental states, and how reactions are regulated by (Boolean combinations of) elemental states.
The regulatory graph is a condensed version of the elemental species-reaction graph, which excludes neutral states (unmodified residues, unbound domains) to improve readability and emphasize information flow.
We find network visualization an indispensable tool in the network reconstruction process.
Parameter-free simulation
Large-scale networks are not only difficult to build and simulate, they are also very difficult to (meaningfully) parametrize. To enable simulation without parametrisation, we develop a bipartite Boolean formalisms that can be used to translate any rxncon model into a ready-to-simulate Boolean model.
The model export is based on two generic update rules that corresponds to the reaction (state updates) and contingency (reaction updates) layers. We define the expected behaviour for two minimal motifs, to create update rules that can be combined - LEGO brick style - into large network models.
The unique mapping of a rxncon network on a bipartite Boolean model makes it possible to validate model structures without parametrisation - a potent tool in network reconstruction that was previously missing for signaling network models. In addition, it allows for qualitative genotype-to-phenotype prediction, down to residue resolution.
Rule-based models
In this project, we develop the semantic mapping form rxncon to a rule based model in the BioNetGen language (bngl). The bngl model can be executed either as an ODE system (for small models) or through agent based simulation in NFsim.
The state of the art in quantitative modeling of (large-scale) signaling models is rule-based model definition with agent-based execution. By defining the system as rules, i.e. reactions for molecule patterns, the model definition becomes scalable. By executing the rules through agent-based simulation the complexity scales with the number of molecules instead of the number of possible states, making execution of large signaling networks feasible.
We implement the translation logic in the rxncon compiler, and explore algorithms for parameter estimation of agent based models.
Modeling cellular regulation
A comprehensive model of the cell division cycle control network
In this project, we formalize biochemical knowledge into a comprehensive mechanistic model of the cell division cycle of baker’s yeast, Saccharomyces cerevisiae.
The cell division cycle is the process through which one cell proliferates into two. It is one of the fundaments of life and highly relevant for health and disease. Cell division is strictly controlled by an intricate signaling system that synchronizes three different duplication events: DNA replication, nuclear division and cell division, hence ensuring ordered duplication and separation. Errors in this regulation may lead to cell death or genomic instability and consequently to severe diseases such as cancer. To understand the function of this system mechanistically in terms of biochemical reactions will be of fundamental importance to understand life - as well as to diagnose and treat diseases due to errors in this control network.
Based on a comprehensive literature review, we encode a detailed mechanistic model in the rxncon language. We use an iterative workflow of literature curation, visualization and bipartite Boolean simulation to build, validate and debug the network model. In the end, we use parameter-free simulation to analyse the predicted phenotype of both deletion and point mutations.
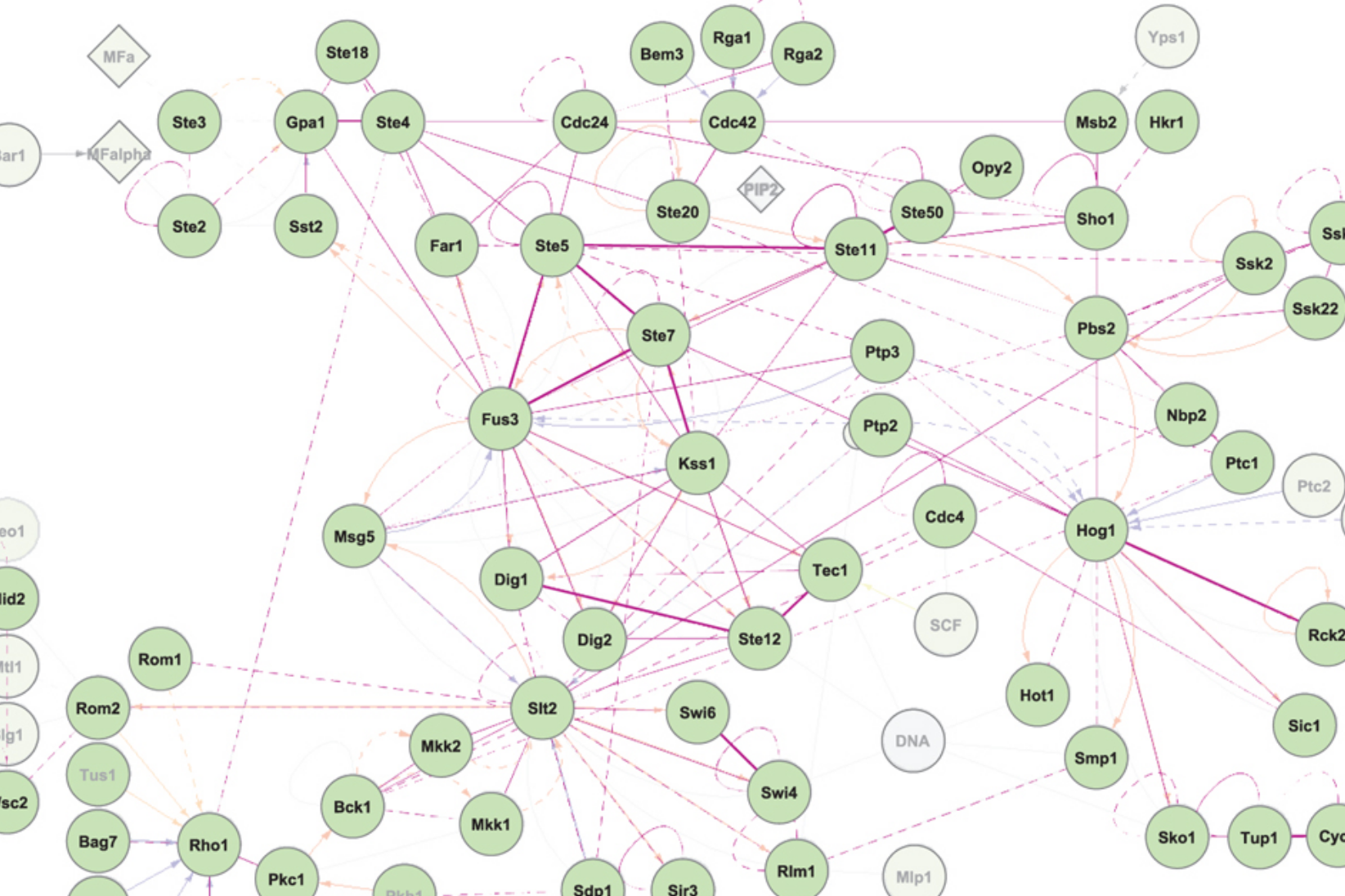
The signal transduction network of baker’s yeast
We are building comprehensive - in scope and detail - models of signal transduction networks in baker’s yeast.
We are following two approaches. First, using an iterative literature curation, visualization and simulation workflow to build, validate and debug the network models. This has yielded highly detailed models of the MAP kinase and Snf1/AMP kinase pathways. Second, we explore the ability to reuse previous curation efforts in the CellDesigner format. To this end, we design a translation algorithm to convert network maps defined in the CellDesigner format into executable rxncon models. The main challenge is to deal with the information content difference: The explicit CellDesigner models lack part of the mechanistic information necessary in rxncon, such as bond topologies in complexes and the identity of the catalytic subunit in catalytic complexes. How to add this information and how to distinguish real information from simplifying assumptions remain outstanding questions.
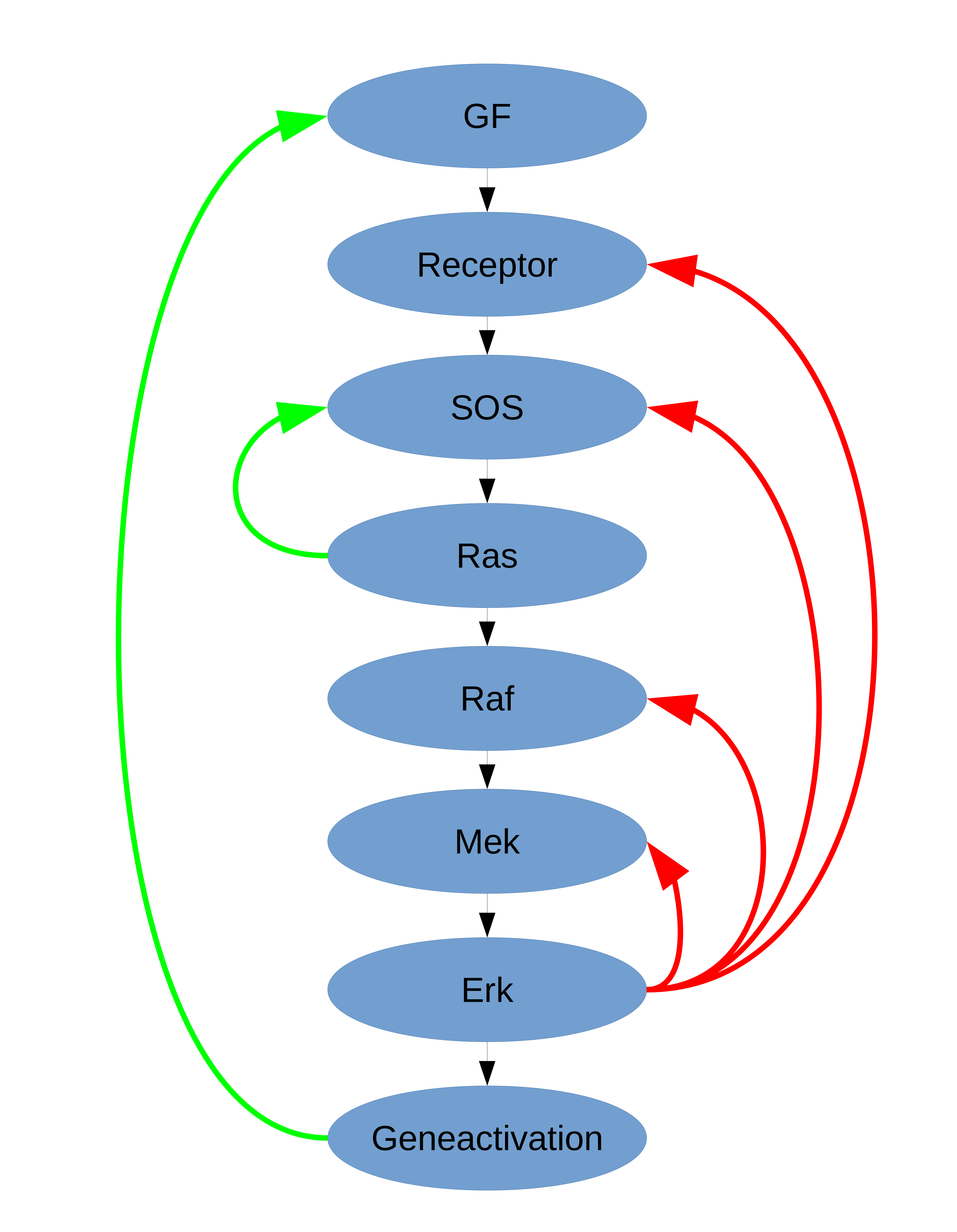
A rxncon model of the RAS/RAF/MEK/ERK signaling pathway
In this project, we explore the possibility to turn the non-executable network maps of the Reactome database into executable rxncon models. To this end, we translate the RAS/RAF/MEK/ERK and Insulin models in the Reactome database into the rxncon language, filter out technical assumptions and validate the resulting models through parameter free simulation.
Modeling tools
The rxncon compiler and workbench
The rxncon compiler is an open source software development project. In the rxncon compiler, we implement the validation and compilation of rxncon models. We currently support three graphical and two executable compilation targets. The rxncon compiler is distributed under the lGPL licence and the code is available at GitHub (Github/rxncon/rxncon). It can be installed through the python package index with “pip install rxcnon”.
We develop the rxncon workbench to lower the threshold for using rxncon. The workbench includes a local model repository and the rxncon compiler under a common GUI. The workbench is coming soon.
A workflow for reconstruction, validation and simulation of large-scale mechanistic signal transduction models
The metabolic modeling community has established a powerful workflow for model building, validation and gap-filling. Here, we develop an analogous workflow for signal transduction, based on tools that can tackle these networks at a large scale.
The workflow uses the rxncon language for model definition, the rxncon reaction and regulatory graphs for visual validation and the bipartite Boolean modeling formalism for computational validation.
PEOPLE
Maria Dost Patrick Segelitz Dr. Marcus Krantz Ulrike Münzner Dr. Jesper Romers Sebastian Thieme Dr. Magdalena Rother Mathias Wajnberg Ulrike Münzner Dr. Timo Lubitz